Introduction
Dentinogenesis imperfecta (DI) is an autosomal dominant genetic disorder characterized by abnormality of the dentin affecting both primary and permanent dentitions in the absence of any other systemic disorder. DI affects 1 in 8000 births and is complicated to manage.[1],[2],[3] There is defect in the structure and composition of the dentin due to an improper production and formation of the collagen bundles resulting in localized form of mesodermal dysplasia of the dentin.[1],[4]
Barret in 1882 termed soft brown, translucent teeth with obliterated pulp chambers without bone disease as hereditary opalescent dentin or Capdepont's teeth.[5],[6] The term dentinogenesis imperfecta was coined by Robert and Schour in 1939[7] and the condition was first described in 1887 in a case involving a "completely normal boy, with dark staining on the teeth, which at age 16 showed abrasion at gum level[8] (Table 1).
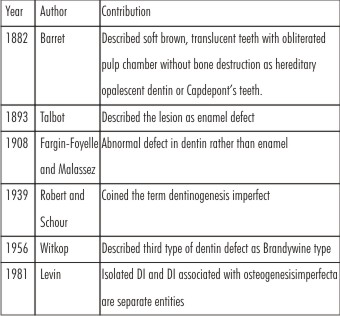 | Table 1: Historical Perspective
 |
Clinically, affected teeth appears yellow to brown in color with translucency. Radiologically, the teeth are characterized by bulbous crowns, marked cervical constrictions with short, thin roots. The pulp chambers and root canals eventually obliterate with the advancing age. Histologically, the thickness and structure of the enamel is normal but the dentinoenamel junction appears smooth and uniform. Dentin presents scarce dentinal tubules that are small and irregular. Occasionally, areas of interglobular dentin are present.[4],[7],[9]
In 1973 Shields, Bixler and El-Kafrawy proposed 3 types of dentinogenesis imperfecta. Type I is defect associated with osteogenesis imperfect, Type II is classical heredity opalescent dentin, Type III was the type found in the Brandywine isolate of Maryland. To overcome the shortcomings of Shields classification a new revised classification was formulated by Witkop which includes Type 1 corresponding to type II of Shields (DI without osteogenesis imperfecta), Type 2corresponding to Type III of Shields (Brandywine type). No substitute for type I of Shields classification was included in this revised classification .[5],[14] (Table 2).
 | Table 2: Classification
|
This case report aims at describing the histomorphological aspects of the DI-II (Shields Classification) and to aware general dentist about this rare genetic disorder.
Case Report
A 19 year old male reported to the department of Oral Medicine and Radiology of our institute with a chief complaint of pain and chipping of teeth. His history revealed that primary and permanent teeth had an unsightly discoloration of the teeth. There was no history of unexplained hearing loss, drug usage, unusual bone brittleness or any other systemic disease in the past or present. There was gradual chipping of both dentitions, may be due to lack of diagnosis thus no proper treatment was provided. On further investigation, he reported that his grandmother had same kind of attrition and discoloration of the teeth.
Intraorally 36, 37, 38, 46 and 47 were missing and all remaining teeth were attrited to the level of the gingiva except third molars. Generalised yellowish brown discoloration with translucent opalescence of all erupted teeth was evident. There was presence of vertical cracks in the enamel structure, chipping of enamel leading to exposure of underlying soft dentin resulting in attrition. Due to attrition loss of vertical dimension was present giving a typical appearance of edentulous patient. (Figure 1) Panoramic radiography revealed bulbous crowned teeth with thin and small roots having marked cervical constriction, prominent in the maxillary molars. Partial or complete obliteration of the root canals was also evident with generalised horizontal bone loss. (Figure 2).
 | Figure 1 : Clinical picture of the patient, a; front view b; right lateral view c.left lateral view d; occlusal view. Note yellowish brown discoloration of teeth
|
 | Figure 2 : Panoramic radiograph showing bulbous crown with cervical constriction and thin roots. Obliteration of the root canals is also evident.
|
Differential Diagnosis include conditions like osteogenesis imperfecta (bone fragility with multiple fractures, blue sclera and hypoaccusis), dentin dysplasia (mostly seen in permanent teeth, shortened roots with crescent pulp chambers and extreme tooth mobility leading premature exfoliation), Tetracycline staining (history of drug usage lead to yellow to dark brown discolouration of affected teeth) and congenital erythropioetic porphyria( autosomal recessive condition due to deposition of porphyrins lead to reddish brown discolouration of permanent teeth).
Based on the clinical and radiological findings provisional diagnosis of dentinogenesis imperfecta II (Shields classification) was made.
Mandibular 2nd premolar which was severally attrited with minimal crown portion was extracted and submitted for histological examination. Macroscopically, tooth was yellowish brown in colour, with rough and attrited occlusal surface. The longitudinal cut section of the tooth was examined under a stereomicroscope which exhibits globular and interglobular dentin with periodic irregular dentinogenesis. There was few disorganised tubular structure with an increased caliber which were more pronounced in the circumpulpal dentin. The pulpal space was obliterated by an unusual type of calcified material consisting of dentin, and deeper layers of dentin had an atypical tubular pattern. (Figure 3).
 | Figure 3 : Stereo zoom pictures of the tooth; note: atypical arrangement of dentinal tubules and obliteration of the pulpal space.
|
Histopathological examination of prepared ground section of tooth revealed short disorganised tubules coursing through an atypical granular dentin matrix demonstrating areas of interglobular dentin formation. The dentinoenamel junction exhibited an irregular scalloped appearance and at places was smooth and uniform (Figure 4), confirming the diagnosis of DI-II based on the clinical, radiographical, histopathological features and lack of evidence of systemic bone disease.
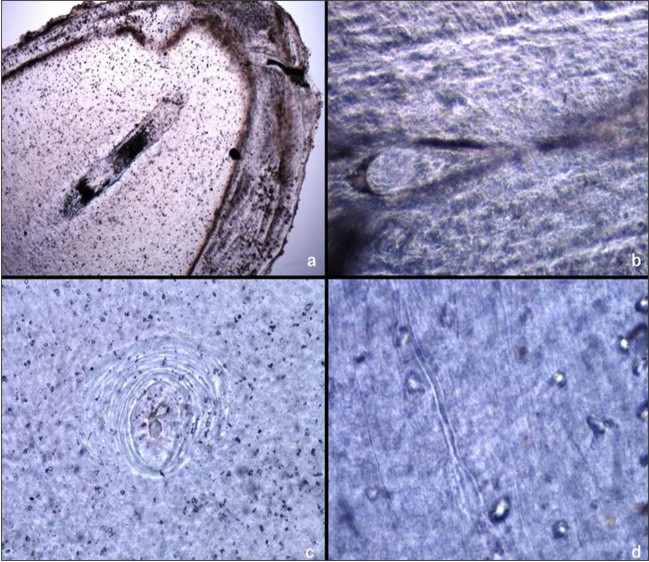 | Figure 4 : Photomicrograph of a ground section of received tooth showing a; radicular remnant pulp b; coronal remnant pulp c; interglobular dentin d; atypical dentinal tubules and granular matrix
|
Discussion
DI is one of the most common disturbances of dentine formation.[10] Sequence of events as a consequence of abnormalities in the dental papilla, the odontoblast dysfunction and impaired terminal odontoblast differentiation leads to abnormal distributions and dimensions of dentin tubuli and disruption of dentin matrix biomineralization and ultimately resulting in DI.[11] DI has been attributed to mutations in the dentin sialophosphoprotein (DSPP) gene, encoding two dentin-specific non-collagenous matrix proteins, dentin sialoprotein (DSP) and phosphoproteins (DPP).[12] DSPP is located within human chromosome 4q22.1 and consists of 5 exons spanning approximately 8343 BP. DSPP is expressed in a number of tissues including bone, kidney, salivary gland and lung but its expression in dentin is hundred times higher than in other tissues. [13]
In DI, the primary teeth are more severely affected than the permanent and it usually affects whites.[15],[6] DI is characterized by dentition with gross abnormality of dentinal tubules and dentinal calcification with normal enamel, cementum, periodontal ligament and a color variation that ranges from gray to brownish violet or yellowish brown, with a characteristic unusual translucent or opalescent hue.[16] The deposit of pigments and minerals in the interior of the dentine tubules alters the colour of the teeth.[10] Excessive wear and fracture of teeth in DI is attributed to cracks and detachment of enamel leading to the exposure of underlying dentin and abnormal dentin-enamel junction that lacks normal scalloping.[16] The dentinal tubules are tortuous, narrow (<1µm), irregular both in shape and size as well as few in number and odontoblastic extensions are found within.[10] They may not penetrate the entire thickness of dentin due to their short length leading to excessive intertubular resulting in transparent dentin.
The tooth morphology is unusual for its excessive constriction at the cementoenamel junction, giving crown a Tulip or bell shaped appearance.[17] In adults, they may frequently wear down to the gingiva. Some patients demonstrate an anterior open bite.[7] DI can be associated with Ehlers Danlos syndrome, Goldblatt syndrome, Schimke immuno-osseous dysplasia, Brachioskeleto-genital syndrome and osteodysplastic and primordial short stature with severe microdontia, opalescent teeth, and rootless molars.[2]
Brandywine isolate was characterized by the presence of unusual pulpal enlargement known as shell teeth. It demonstrates normal thickness enamel in association with extremely thin dentin and dramatically enlarged pulps. The thin dentin may involve the entire tooth or be isolated to the root. This abnormality has been seen most frequently in deciduous teeth.[18]
Radiographically, the crowns are bulbous with a constricted area at the cementoenamel junction. The roots appear shortened and conical or ‘spike like.’ In both primary and permanent dentitions the pulp chambers and root canals are often obliterated.[3] The short roots and bell shaped crowns are also obvious on radiographs. In type II (Brandywine type) the dentin appears thin and pulp chambers and root canals are extremely large giving appearance of thin dentin shells-hence the name shell teeth.[17] DI-2 differs from DI-1 by the presence of multiple pulp exposures, normal non mineralized pulpal space and a general appearance of “shell teeth”. [14]
Histologically, DI is typified by dentin with irregular tubules, pulpal obliteration, interglobular dentin, large areas of uncalcified matrix.[6] The dentin in the external part shows proper structure however in the para-ventricular part it is improper with the canals of a disturbed shape and course; also it has got irregular inter-spherical spaces.[19],[20] Scanty atypical odontoblasts line the pulp surface and cells can be seen entrapped within the defective dentin.[18] The histological appearance of the enamel is reported to be normal although hypocalcified defects may be present. The dentinoenamel junction lacks the usual scalloped topography.[4] Enamel cracking appears within the enamel itself or along the dentinoenamel junction.[3] In DI, water content of teeth is greatly increased as much as 60% of the normal while the inorganic content is less than that of normal dentin that takes its hardness close to cementum, thus explaining the rapid attrition of affected teeth.[14]
Treatment of DI is very difficult and its main purpose is to prevent loss of enamel and dentin due to abrasion. Several types of treatment of DI have been described in the literature modified according to each case. Crowns can be used as a preventive device for the abrasion of the dental structure, can be used in deciduous and young permanent posterior teeth. Shafer et al. (1985) emphasize that restorations cannot be permanent, owing to the low hardness of the dentin. Consequently, when fractures occur at the gingival level or below the gum, exodontia is indicated, as in the case of teeth which exhibit periapical rarefaction and root fracture. [21]
Conclusion
DI results from deposition of defective dentin resulting in yellowish brown discoloration of the dentition with bulbous crowns, short thin roots and obliterated pulpal space. Patients with DI shows wear of tooth structures leading to multiple pulp exposures. This case report is an attempt to create awareness among general dentist about this rare genetic disorder. This will enable early diagnosis and proper treatment thus preventing loss of the dentition and preserve function and aesthetics of the patient.
References
1. Shetty N, Joseph M, Basnet P, Dixit S. An integrated treatment approach: A case report for dentinogenesis imperfecta type II. Kathmandu University Medical Journal 2007;5(18): 230-33.
2. Pai A, Prasad RS, Ramakrishna & Rao, R. Capdeponts teeth - a hereditary dentin defect. Case report & review. Int J Odontostomat 2012;6(2):229-34.
3. Millet C, Viennot S, Duprez JP. Case report: Rehabilitation of a child with dentinogenesis imperfecta and congenitally missing lateral incisors. European Archives of Paediatric Dentistry 2010; 11(5): 256-60.
4. Delgado AC, Ruiz M, Alarcón JA, González E. Dentinogenesis imperfecta: the importance of early treatment. Quintessence Int 2008;39:257–63.
5. Witkop CJ Jr. Amelogenesis imperfecta, dentinogenesis imperfect and dentin dysplasia revisited: problems in classification. J Oral Pathol1989;17:547-553.
6. Subramaniam P, Mathew S, Sugnani SN. Dentinogenesis imperfecta: A case report. J Indian Soc Pedod Prev Dent 2008;26:85-7.
7. Kamboj M, Chandra A. Dentinogenesis imperfect type II: an affected family saga. J Oral Sci 2007;49(3):241-44.
8. Mayordomo FG, Estrelu F, Aldecua EA. Dentinogenesis imperfecta: a case report. Quintessence Int 1992; 23:795-802.
9. Lewis DM. Dentinogenesis imperfect. ODA Journal 2007; 24-7.
10. Leal CT, Martins LD, Verli FD, de Souza MAL, Ramos-Jorg ML. Case report: clinical, histological and ultrastructural characterization of type II dentinogenesis imperfecta. European Archives of Paediatric Dentistry 2010; 11(6):306-9.
11. De Coster PJ. Dentin disorders: anomalies of dentin formation and structure. Endodontic Topics 2012;21:41–61.
12. Knezevic A, Tarle Z and Panduri V. Esthetic reconstruction of teeth in patient with dentinogenesis imperfecta – a case report. Coll. Antropol.2006;1:231–34.
13. Barron MJ, McDonnell ST, MacKie I, Dixon MJ. Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet Journal of Rare Diseases 2008, 3:31.
14. Rajendran R, Sivapathasundharam B. Shafer’s Textbook of Oral Pathology, ch1, 6th ed, Elsevier, New Delhi 2009: 54-55.
15. Majorana A, Bardellini E, Brunelli PC, Lacaita M, Cazzolla AP, Favia G. Dentinogenesis imperfecta in children with osteogenesis imperfecta: a clinical and ultrastructural study. International Journal of Paediatric Dentistry 2010; 20: 112–18.
16. Bhandari S, Pannu K. Dentinogenesis imperfect: a review and case report of family over four generatins. Indian J Dent Res 2008; 19 (4): 357-61.
17. Jindal MK, Maheshwari S, Verma R, Khan MT. Comparative study of dentinogenesis imperfecta in different families of the same topographical region. International Journal of Clinical Pediatric Dentistry; 2009;2(3):27-34.
18. Neville BW, Damm DD, Allen CM, Bouquot JE. Oral & Maxillofacial Pathology, ch 2nd, 3rd ed, Elsevier, New Delhi 2009:106-8.
19. Malmgren B, Lindskog S. Assessment of dysplastic dentin in osteogenesis imperfecta and dentinogenesis imperfecta. Acta Odontol Scand 2003; 61: 72-80.
20. Neeti B. Dentinogenesis Imperfecta – “A Hereditary Developmental Disturbance of Dentin”. The Internet Journal of Pediatrics and Neonatology. 2011;13(1).
21. Modesto A, Alves AC, Vieira AR, Portella W. Dentinogenesis imperfecta type II: case report.Braz Dent J 1996; 7(1):47-52.
|